Never use re-docking for estimation of docking accuracy
Re-docking of ligands found in PDB files are often used as a fast evaluation of a docking program before working with designed or other ligands. However this re-docking can give deceptively good results and it is recommended to instead test with cross docking. In this follow up blog post I give a short example of how it can be done.
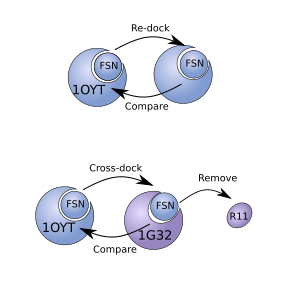
Re docking vs. Cross docking
Cross docking with Smina
Last time I illustrated how easy it is to use Smina for docking a small molecule ligand to its receptor. However, the excellent results can be deceptive if only re-docking is used for estimation of the docking accuracy. Re-docking is the process of removing the ligand molecule from the receptor model and then dock it back. Instead cross-docking should be used. Here a receptor model obtained from a PDB file where another ligand is removed is used for docking of the small molecule. Its easy to download a PDB file, remove the ligand and compare with the original, but as I will show, the results can be much different, when the much more cumbersome procedure of cross docking is used.
The two different procedures are conceptually outlined in the figure to the right. The PDB file with ID 1OYT was time downloaded, the ligand (id: FSN) removed and re-docked. This is what was done in the previous blog post. For cross docking shown below, the 1G32 receptor will be, aligned to the receptor model in 1OYT, the 1G32 ligand with ID R11 removed and the receptor used for docking.
Receptor file download, alignment and saving is easily done with a script for PyMOL:
fetch 1OYT fetch 1G32 align 1G32, 1OYT remove resn HOH h_add elem O or elem N #Select select 1G32-R11, resn R11 select 1OYT-FSN, resn FSN select 1OYT-receptor, 1OYT and not 1OYT-FSN select 1G32-receptor, 1G32 and not 1G32-R11 #Save receptors and ligands save 1G32-R11.pdb, 1G32-R11 save 1OYT-FSN.pdb, 1OYT-FSN save 1OYT-receptor.pdb, 1OYT-receptor save 1G32-receptor.pdb, 1G32-receptor
As last time OpenBabel is used to convert to PDBQT file format.
obabel 1G32-receptor.pdb -xr -O 1G32-receptor.pdbqt obabel 1G32-R11.pdb -O 1G32-R11.pdbqt obabel 1OYT-FSN.pdb -O 1OYT-FSN.pdb1t obabel 1OYT-receptor.pdb -xr -O 1OYT-receptor.pdbqt
The align command in Pymol specifies the mobile target first, so the files generated in the previous blog post could actually have been reused as the atom coordinates of the 1OYT receptor did not change. With the PDBQT files generated it is easy to cross-dock one ligand to the other receptor and afterwards in PyMOL compare the result with the original pose of the ligand.
smina.static -r 1G32-receptor.pdbqt -l 1OYT-FSN.pdbqt --autobox_ligand 1OYT-FSN.pdbqt --autobox_add 8 --exhaustiveness 16 -o FSN-Crossdock.pdbqt pymol FSN-Crossdock.pdbqt 1OYT-FSN.pdb 1G32-receptor.pdb 1OYT-receptor.pdb
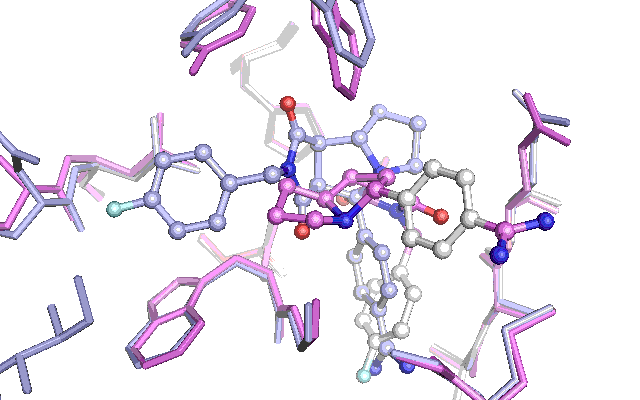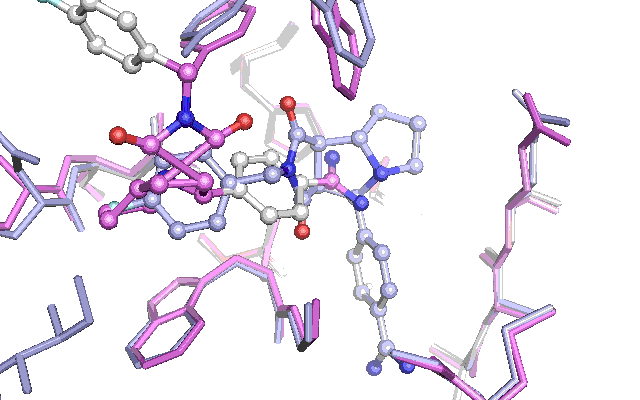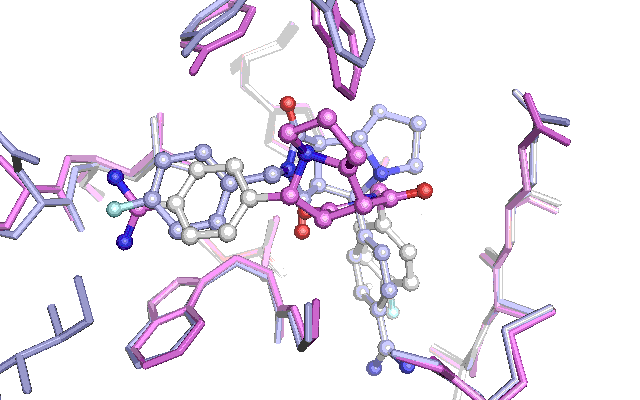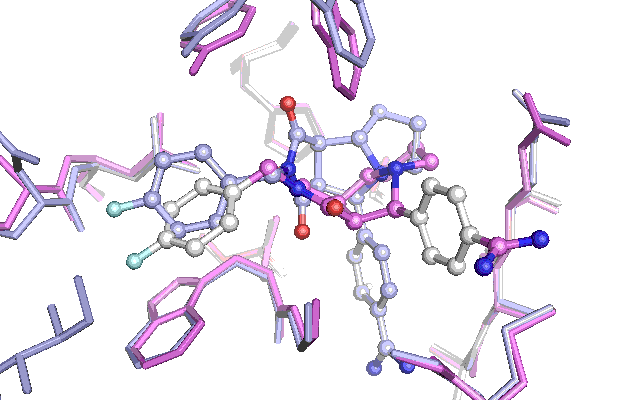
Cross docking animation of top nine poses
The results are not as convincing as last time. The first nine suggested poses fails to find the native pose. The reason seem to be the changes in a tyrosine and tryptophan side chain, which have adapted their position in an induced fit with the other ligand which had been used to produce the Xray structure in the PDB file with the id 1G32. This leads to a steric clash with the native pose of the ligand for the test docking, which penalizes the pose in the scoring function. To get around this a lot of preparation of the docking target is necessary or alternatively the scoring function can be adjusted. Let me know if theres interest for this subject by commenting below.
Docking the ligand from 1G32 into the receptor from 1OYT gives better better results, although not as nice as if the 1G32 receptor is used. So always evaluate docking targets with cross docking if it is possible.
smina.static -r 1OYT-receptor.pdbqt -l 1G32-R11.pdbqt --autobox_ligand 1OYT-FSN.pdbqt --autobox_add 8 --exhaustiveness 16 -o R11-Crossdock.pdbqt pymol R11-Crossdock.pdbqt 1G32-R11.pdb 1G32-receptor.pdb 1OYT-receptor.pdb
Happy docking 😉
Esben Jannik Bjerrum
Pingback: Machine Learning optimization of Smina cross docking accuracy | Wildcard Pharmaceutical Consulting
Hello Esben! can you please comment the paper from Dr. Koes “Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise”. They find there re-docking to give better results than cross-docking.
Thank you for asking. Yes of course! Re-docking nearly always give better results than cross-docking. Re-docking is a much simpler task than cross docking, as the protein is already in an induced fit to the ligand. Whereas in cross docking, the differences in the precise side chain and/or backbone conformations makes it a much harder task to distinguish correct poses from spurious ones. Benchmarking your docking accuracy performance with re-docking will thus lead to overly confidence in the performance of the docking protocols and software. Unless of course you want to re-dock. But what is the practical use case for re-docking?
The docking score function can be re-tuned for better cross-docking performance, as I’ve written about in another blog post. and publication.
What do you plan to use Vina for?
What do you do when the re-docking cannot reproduce the native pose? Is there something doubtful in the crystal structure?
If your docking procedure can’t even redock, there’s something really wrong, and you should try to investigate why the docking fails. It’s difficult for me to say without more details. But check the settings of the docking program? Format of input files incl. evt. atom type assigning and/or charges? But also the PDB structures are not always good models, missing sidechains, flips of his, asn, gln, protonation states, alternate conformations, high b-factor etc. etc. Also, structural waters can sometimes be crucial, evt. check their position and possible binding energy with tools such as GRID from moldiscovery or watermap from schrodinger. And some binding pockets are just hard targets, typically open/surface exposed and with only a few specific interactions. Hope it helps
The question is: did we solve the redocking problem already ?
Thanks for commenting on my old blog post. No, and it has actually been detrimental to pursue and optimize towards an irrelevant metric.
Hello Esben, while cross docking, do we need to have dock in the protein of same species or it can be done to a different species. In your case both the proteins are of Homo sapiens and Hirudo Medicinalis.
Good question and point, it ideally should be the same protein with different ligands, so it’s the same binding site but with different induced fits. But if the binding site is conserved and the proteins have high homology, it could probably work as an estimate. How big differences are there in sequence for the two recombinant proteins in the PDB entries 1OYT and 1G32?