Deep Chemometrics: Deep Learning for Spectroscopy
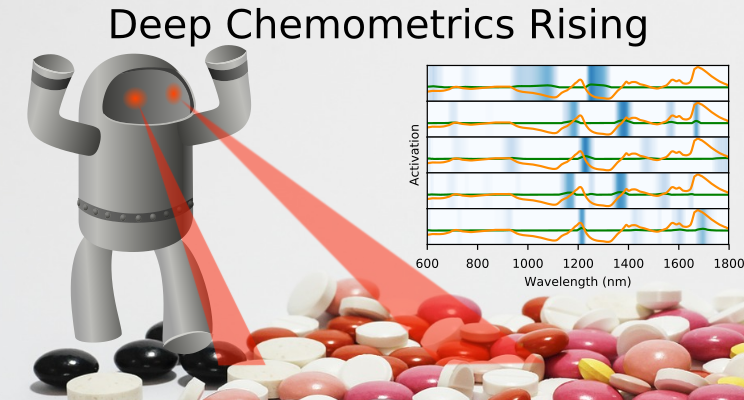 During my postdoc project at the Chemometrics and Analytical Technology section at Copenhagen University I worked with modeling of spectroscopical data with PLS models. Chemometrics is “the science of extracting information from chemical systems by data-driven means” according to Wikipedia, but I have mostly encountered the term when modeling spectroscopical data to predict chemical constitution or contents. With machine learning is is possible to take a cheap and robust spectrocopical measurement of, say a process intermediate, and immediately predict the outcome of a hour long laboratory analysis. In a lot of situations this is rather useful.
During my postdoc project at the Chemometrics and Analytical Technology section at Copenhagen University I worked with modeling of spectroscopical data with PLS models. Chemometrics is “the science of extracting information from chemical systems by data-driven means” according to Wikipedia, but I have mostly encountered the term when modeling spectroscopical data to predict chemical constitution or contents. With machine learning is is possible to take a cheap and robust spectrocopical measurement of, say a process intermediate, and immediately predict the outcome of a hour long laboratory analysis. In a lot of situations this is rather useful.The most commonly used model seem to be PLS (partial least squares). Its a stepwise linear model that extract latent variables and seem to fit very well with a lot of spectroscopical problems. We know from Lambert Beer’s law that there is a linear relationship between a chemical compounds concentration, the path length and the absorption of light at the wavelengths where the compound absorps light, which may be reflected in the good fit to the PLS models.
Going deeper with CNNs
However, after learning more about the use of deep learning and seeing the large success of novel neural network architectures like convolutional neural networks in fields like image analysis and classification I became interested to see how that was applied to chemometrics and analysis of spectroscopical data. To my surprise there didn’t seem to be a lot of applications and literature about it, except for a few stray papers. This is interesting as it seems obvious to leverage the progress in deep learning from analysis of images (2D) to spectral data which can intuitively be seen as 1D images. I discussed this with my supervisor, Thomas Skov, from the postdoc project and we thought this could be an interesting subject for a Masters project to explore the area. Due to my curiosity and impatience I did a few experiments on public datasets and saw promising results and a wildcard intern Mads Glahder, looked further at it. We collected our results in a preprint which is available at http://arxiv.org/abs/1710.01927 where we used convolutional neural networks (CNNs) for the prediction of drug content in pharmaceutical tablets from NIR spectrums.
The study not only showed very good performance of the CNN models. We had to “cheat” to get the PLS models to perform at the same level by assuming that we could predict the perfect number of latent variables. It was amazing to see how robust the CNN models were to over fitting. The use of the convolutional architecture efficiently limits the number of weights and parameters in the first layers, which probably smoothed out noise and help counteract over fitting even though the entire model contained millions of weights/parameters.
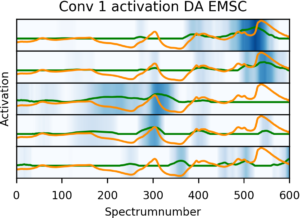
Activations of the five most active kernels (neurons) in the first convolutional layer when analyzing the reference spectrum (orange), the activations is shown as the blue shades and the green curve.
The activations of the kernels (different mini networks in the first convolutional layer) enabled us to get a better qualitative understanding of the way the network works. The transformations done by the kernels had some resemblance to known spectral preprocessing steps such as derivatives, smoothing and variable selection used for enhancing the performance of PLS models. This was a nice link back to known chemometric territory, but the preprocessing steps was entirely “reinvented” by the network itself to solve the given data analysis and prediction task. The final network we used was rather shallow compared to the current state of the art image classifiers, but expanding the datasets or compiling more data into multi task networks would probably enable more varied and relevant kernels to be developed in even deeper networks.
Spectral data augmentation and EMSC
A lot of efforts have gone into developing preprocessing methods to be employed before the PLS modeling. One example is multiplicative scattering correction (MSC), which is a technique for baseline correction. In deep learning the strategies seem to be completely opposite. Here noise and variation is artificially introduced to in the available data material to get more training samples from the limited number of labeled samples. We did this by simulating the baseline variations and noise, so that the network can better handle the variations in the future. Introducing noise and variation seems like the completely opposite strategy than trying to eliminate the baseline variations via MSC. PLS models didn’t benefit a lot from the data augmentation (in fact they seem to get worse), but the neural network models did improve. To our surprise the best preprocessing for the neural network was actually to first simulate the baseline variations and then to remove it again with MSC. More close investigation showed that there were residuals of the variations left after the MSC correction of the data augmented samples, which may explain the improvement as the neural network could get the best of two worlds. I think that it is beautiful. It somehow seems reassuring that it is the combination of the traditional domain specific methods with the new approaches from deep learning that gives the best performance. Domain knowledge is probably not going to be completely replaced by “blind” data science and machine learning in the near future.
You can read the details in the preprint publicly available at http://arxiv.org/abs/1710.01927
Example Code
Some scikit-learn like utilities for spectroscopical preprocessing and two example example jupyter notebooks on how to data augment, build and train the Keras models are available on the GitHub repository: https://github.com/EBjerrum/Deep-Chemometrics
Future Perspectives
There is a lot to research and try out! Can we improve generalization of the kernels by expanding the dataset for multi task learning? Do more advanced architectures like ResNets, Inception modules, GAN architectures etc. etc. give even further benefits? Does it work equally well on all kinds of spectroscopical datasets or is there spectroscopical domains where they excel and others where CNNs is a bad choice? Interested students should take a look at http://food.ku.dk/english/education/project-proposals/details/?obvius_proxy_url=http://www2.bio.ku.dk/cms/projekter-food/detaljer.asp?ID=48
To summarize our findings:
Pros:
- Excellent extrapolation with regard to compound content and across instruments
- A novel spectral data augmentation technique improved predictive performance
- An initially counter-intuitive combination of spectral data augmentation and EMSC was the best data preprocessing option.
- Robust against over fitting despite high number of variables
- Better or on par performance than a baseline PLS model
(even though we assumed we could predict the optimal number of components for the PLS model)
Cons:
- A bit more lengthy training
(15 minutes on consumer GPU)
- Less interpretable than PLS
Cheers
Esben Jannik Bjerrum