Better Deep Learning Neural Networks with SMILES Enumeration of Molecular Data

The process of expanding an otherwise limited dataset in order to more efficiently train a neural network is known as Data Augmentation For images there have been used a variety of techniques, such as flipping, rotation, sub-segmenting and cropping, zooming. The mirror image of a cat is still recognizable as a cat, and by applying the mirror operation the neural network will get more examples to train on.
But how can this be done for a molecule? The mirror image may be the enantiomer, but that may not be what we want? For SMILES there exists the possibility of enumerating the different possible SMILES, which was the subject of my preprint publication at arxiv.org: SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules
SMILES enumeration for data augmentation
Simplified Molecular Input Line Entry System (SMILES) is a brilliant molecular data format, which can uniquely encode the structure of a molecule in a single line using standard text characters. “C” denotes an aliphatic carbon, c is an aromatic carbon. numbers starts and closes a ring, so c1ccccc1 is a six membered ring of aromatic carbons (a.k.a. benzene). Branching is encoded by parentheses. Double and triple bonds can be specified with = and #, cis-trans isomery around a double bond can be shown with /C=C/ or /C=C\. Even stereo chemistry can be encoded with @ and @@. Because it is a text format it is easily used in standard computer programs such as spreadsheets and documents.
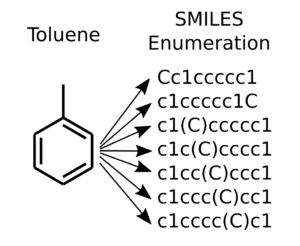
SMILES enumeration of toluene, the topmost is the canonical SMILES
For really simple molecules such as methane “C” or ethane “CC”, there is only one SMILES string. (Unless of course the implicit single bond of ethane is specified “C-C”). Already with propane, there exists two possibilities for using branching “CCC” and “C(C)C”. This depends if we start the SMILES from the center atom or the from one of the ends. Toluene has seven different ways of representing the branching as illustrated in the Figure to the right.
Canonical SMILES is a unique way of writing a SMILES for a molecule, where some rules about numbering defines the ordering of the atoms. This ensures that there is only one unique SMILES corresponding to one unique molecule. It is often useful to have this 1:1 correspondence. However, for data augmentation purposes it is beneficial to go the other way. This way a limited size molecular dataset can be significantly expanded for extra training samples.
But does it work?
The simple answer is: Yes it appears so. In the preprint paper I used a molecular dataset with IC50 dataset of DHFR inhibitors. Here a clear improvement was observed in the neural network prediction of a test set. It was possible by careful tuning to build a model using only the canonical smiles, but it was much easier to build a model with the expanded data set. The model using the expanded dataset was moderately better at predicting the test set of either enumerated or canonical SMILES. The model that had been build using the canonical smiles were slightly worse at predicting the test set with canonical SMILES, but failed miserably at predicting a dataset with enumerated SMILES. The best prediction could be obtained by predicting the IC50 from a compound using all its enumerated SMILES and then take the average, where the correlation coefficient was 0.68 and the RMS was 0.52. In comparison, the model trained on canonical smiles and predicting canonical smiles obtains a correlation coefficient of 0.56 and a RMS of 0.62.
How do you build a QSAR model directly from SMILES?
It can appear surprising that it is possible to use SMILES directly, but the trick is to use LSTM cells in batch mode. LSTM cells for recurrent neural networks have been covered before on this blog Teaching Computers Molecular Creativity. In batch mode the final state of the LSTM cells after iterating through all the characters of the SMILE are fed forward in the neural network, which can be a standard feed forward neural network with a single output, which should predict the IC50. The architecture is illustrated in the figure.
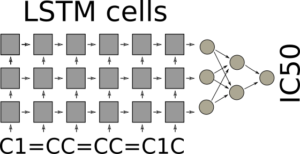
LSTM network for QSAR modeling
The LSTM cells are very good at keeping their internal state, so as a highly speculative example: One cell may remember that it saw a 1 directly after a lower case c, another may count the numbers of lower case c’s after a number, and if this matches five and a new number 1 is encountered, a third cell, the “Benzene” cell is switched on. By cooperation the cells have recognized a benzene ring. If this is exactly how it actually happens I don’t know. Often it can be quite difficult to understand whats happens inside the neural network with its enormous amount of connections, even though sometimes a cells function can be understood. During training of the network, the end state of the LSTM network should support the prediction of the property of interest and it could be that the network learns to recognize the molecular features in the SMILES strings that are most suitable for the task at hand.
Is it better than using descriptors or fingerprints?
I don’t know yet…. I’m still investigating the issue. It seems to be on par with the performance I can get from optimized models using fingerprints. Below is a comparison of a LSTM-QSAR model trained on enumerated SMILES and a QSAR model built using standard molecular fingerprints. The dataset is from the TOX21 challenge, which I have used before: Tune your deep tox neural network with free tools. The ROC-AUC seems to be very similar, so the predictive performance is supposedly the same. It may be that the performance is in fact rather limited by the dataset than how we exactly analyze it. Anyway, It’s a new and different way of reading molecular information into neural networks.

Comparison of LSTM-QSAR modelling using SMILES as input vs. a traditional Neural Network modelled using molecular fingerprints
If you happen to download the preprint and have some comments or suggestions, please let me know as I’m not finished with the investigations. Please comment below.
Best regards
Esben Jannik Bjerrum

CEO, Esben Jannik Bjerrum
Very cool work.
I am thinking of implementing your method, although it is not a high priority for me right now. I work with compounds that have a lot of nitro and azide groups.
Interestingly there are three ways to represent nitro groups: [N+]([O-])=O or [N+]O=[O-] or N(=O)=O (the ” expanded valence representation” proposed for canonical SMILES, see https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3495655/)
There are at least two ways to represent azides as well. For both nitro and azides this is all due to the fact they are resonance structures.
Regards,
– Dan
Thanks. You should normalize your compounds in the training/dev/test sets to your wanted nitro-group representation. The model should then learn your wanted representation. Btw, [N+]O=[O-] looks a bit strange to me.
Hi, yes, sorry I messed up the SMILES string there, it should be [N+]([O-])=O.
I tried to run this part of the code : https://github.com/Ebjerrum/SMILES-enumeration#batch-generation-for-keras-rnn-modeling (with the example dataset).
When I run ‘X,y = generator.next()’ i get this error message:
File “SmilesEnumerator.py”, line 181, in transform
for i,ss in enumerate(smiles): KeyError: ‘C’
When I run it again, I also get KeyErrors for ‘n’ and ‘c’.
Any idea how I can fix this?
Hi, sorry for the late reply, but you comment was marked as spam. Key errors are usually arising because the list of characters for the vectorization is not complete. The .fit() method takes an array of SMILES strings and builds a character set from that and calculates the max length. Did you use this before trying to use the generator?
But sometimes thats not enough. When enumerating the SMILES strings, the number of cycles can become greater than in the original dataset, and some extra chars can appear. Therefore there’s the extra_chars option to the fit method.
When you are missing C and c and n, theres something seriously wrong though. have a look at the .charset property, what does that contain? The default should contain the C,c and n.
How is PC_uM_value derived?
I assume you talk about the DHFR dataset. Its encoded as a tag in the SDfile. Needs some cleaning though, as it’s text.
Pingback: A simple LSTM based QSAR model in PyTorch | Cheminformania
What exact part of the code generates multiple SMILES for a given SMILES. In the readme file, a for loop has been used to demonstrate the working of the randomize_smiles method, however while using the generator in Batch generation for Keras RNN modeling, I don’t understand how the looping is being done and what is the number of augmented SMILES generated per sample.
Hi, Thanks for commenting. The code generating the SMILES for a batch is line 174-194 in https://github.com/EBjerrum/SMILES-enumeration/blob/master/SmilesEnumerator.py. Here an empty numpy array is created with the dimensions batch, sequencelength, charset, and then itereatively filled with the enumerated SMILES from the batch. One SMILES in the batch will be augmented one time. However, next epoch a new SMILES form will be generated. As the number of possible SMILES often are much larger than the usual number of train epochs, you’ll effectively prevent overfitting on the specific SMILES (but can still overfit on the molecules as such).
Also, please do check up the more general and slightly more up-to-date molvecgen package: https://github.com/EBjerrum/molvecgen, which amongst others contain SMILES augmentation code.
We’re preparing another framework which are pytorch oriented and will soon be released.
Thank You for the clarification!