rdEditor: An open-source molecular editor based using Python, PySide2 and RDKit
At the RDKit UGM 2018 in Cambridge I made a lightning talk where I show cased rdEditor. I’ve wanted to write a bit about it for some time. The project started as I had a need to annotate some atoms of molecules in a dataset, but couldn’t really find a good Python based tool to customize for my needs. After my first prototype that helped me annotate the dataset I took the basic solution where I could click on a RDKit depiction and make an action on the clicked atom and turned it into a more general purpose molecular editor.
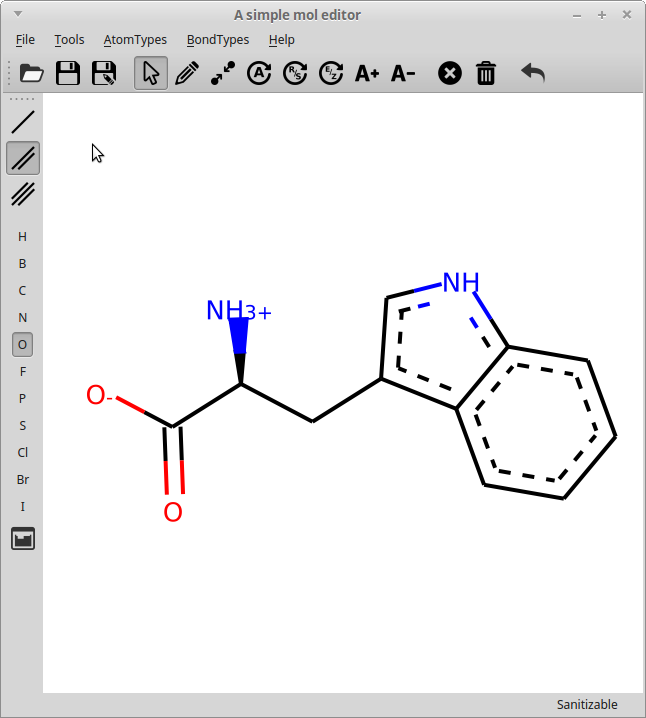
rdEditor main window
The editor can load and save .MOL files and allows editing the molecule, including:
- Select atoms
- Adding new atoms and bonds
- Create bonds between existing atoms (make rings)
- Edit atom and bond types
- Delete atoms or bonds
- Toggle stereo chemistry (both R/S and Cis/Trans double bonds)
- Increase and decrease formal charges
It’s and editor, not a drawing tool
I’ve avoided to make a drawing tool which gives some pros and cons. On the up side, the tool ended up being simple to code, as I got all the chemistry knowledge from RDKit and could reuse the drawing capabilities of RDKit directly. The chemistry is RDKit controlled as the RDKit molecule object is manipulated directly, so bond lengths and atom and bond types are handled by RDKit. On the downside, by avoiding the idea of a “canvas”, the recalculation of the 2D coordinates from the RDKit molecule can make the molecule a bit “Jumpy”. Especially when the molecule is small. After a suggestion from Greg Landrum at the UGM, some code was added at the hackathon that reuses existing coordinates if they are present and only recalculates the added atoms. This minimized but didn’t completely remove the “jumpiness”. Additionally, the molecular layout can’t be user controlled, which may be an issue if one wants to make pretty pictures of molecules. However, there’s already plenty of tools for that .
The Code
The editor is based on PySide2, a project very similar to PyQT, but with another licensing scheme (LGPL >< GPL/Commercial Licensed). QT is the User interface from Trolltech and PySide and PyQT are python bindings to the C++ libraries. They are VERY similar in api, so most code can switch from PyQT to PySide by simply importing the other bindings instead with maybe some slight modifications. The rdEditor code itself is entirely Python based and ~1000 lines of code, so it should be fairly easy to customize if there is a need. I’ll not show all the code here, but just write a bit about the organization and maybe highlight some pieces of it.
The Widgets
The code is organized into a couple of modules. As show in this figure:
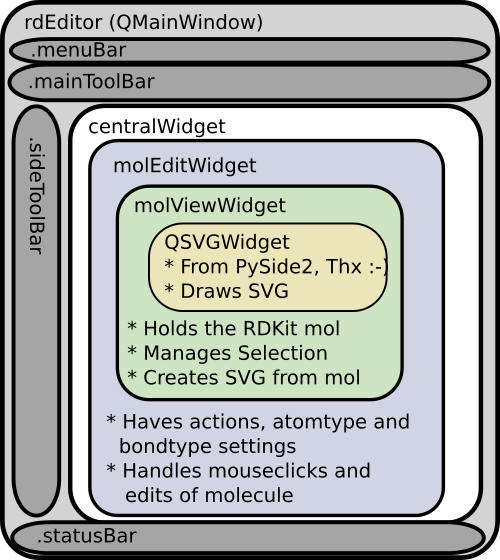
We’ll start from the central part. The MolViewWidget is the fundamental widget and its purpose is to provide a PySide2 widget that can display an RDKit molecule. It’s a subclass of the PySide QSVGWidget which shows SVG, and have been customized to handle a .mol property and update the drawing if molecule itself is changed. The .mol property on the widget is coded as a property with getter and setter functions. The getter simply returns the hidden ._mol property, but the setter check the equality of the molecule, updates the hidden property and emit the QT signal molChanged. This QT signal is then bound to the function sanitize_draw as a slot “self.molChanged.connect(self.sanitize_draw)”. I’ve previously written a bit more extensively about signals and slot used in a simple SDfile browser. Putting the automation into the setters and getters makes it easy to set the molecule and get it shown. The only thing needed to change the molecule other places in the code is:
viewwidget.mol = Chem.MolFromSmiles('CCc1ccccc1N')
and the widget will prepare and display the molecule. The signal “molChanged” can also be bound to other slots in other pieces of code if additonal actions are needed. The getter method also make a copy of the molecule in the _prevmol property to support the undo function.
The molViewWidget also has some code to handle “selections” of the molecule. It’s basically a list of selected atom index numbers for the current molecule, with some functions to add or remove existing atom index numbers. Changing the selection will force a redraw, but not a recalculation of the atom coordinates, and the atoms will be highlighted with the latest selected atom marked with a darker red.
The molViewWidget should be easily reusable in other PySide projects, such as browsers. Programming a simple molecular G indicesUI browser with model-view architecture (MVC) using Python with PySide or PyQt and RDKit. The linked example doesn’t use the widget though, but is a complete walkthrough.
The molEditWidget is a further subclass of the molViewWidget and adds functions that handles clicks on the canvas and methods that manipulate the RDKit molecule. The mousePressEvent from the original QSVGWidget has been overloaded, and from the click event, the SVG coordinates clicked are extracted. These are then converted back into RDKit molecule coordinates and the distance to the nearest bond or atom is calculated.
The conversion of the click to RDKit molecule coordinates were a bit tricky. The SVG drawing code of RDKit rescales the 2D coordinates into the SVG coordinates in the drawing and after drawing its possible to use the RDKit drawer.GetDrawCoords() function to get the drawing coordinates from molecular coordinates. But there was seemingly no function to go in the other direction. The scaling factors which are probably stored at the C++ level is also not exposed. The workaround was to feed the GetDrawCoords() method two rdkit.Geometry.rdGeometry.Point2D points (0,0) and (1,1) and then calculate the scaling and offset from the returned SVG coordinates.
def SVG_to_coord(self, x_svg, y_svg):
if self.drawer != None:
scale0 = self.drawer.GetDrawCoords(self.points[0])
scale1 = self.drawer.GetDrawCoords(self.points[1])
ax = scale1.x - scale0.x
bx = scale0.x
ay = scale1.y - scale0.y
by = scale0.y
return Point2D((x_svg-bx)/ax, (y_svg-by)/ay)
else:
return Point2D(0.,0.)
If the drawer is not present, there is not yet defined any atoms and the function returns the origin. To go from the clicked coordinates to the atom or bond, the clicked coordinates are compared with lists of atom coordinates and bond coordinates (their centers). If the minimum distance found is within a given threshold, the atom or bond is clicked, otherwise the background is clicked.
Determining the action
The molEditWidget have three properties that determines what should happen after the click. There are chemistry properties “.atomtype” and “.bondtype”, that determines what atom and bondtype the action should be performed with and a “.action”, that determines what action should be performed. The combination of the properties and the type of click, atom, bond or background, determines what method to ultimately call. As an example; if the .action is “replace” and the .atomtype is “N” or 7 and an atom is clicked, the atom type in the molecule will be updated. Of course not all combinations of actions and atom/bond types makes sense.
Toggling bonds
To give a the possibility to “toogle” bond types, the combination of “add” and a bond-click is dispatched to the toggle_bond function. Here the bondtype of the clicked bond is used to lookup the next by comparing with a list of standard RDKit bond-types and selecting the next in list. It is still possible to use the replace action with the bondtype with all other bond-types defined in RDKit from the menu. The bondlists starts with the last item duplicated, so that it will start over if that bond-type is the current one. If the previous bondtype is not in the list, the argmax will return 0 and thus select the next in the list, which is single. The final line triggers the QSignal “molChanged” and all functions that are bound to this will be executed (redrawing the molecule etc.).
def toggle_bond(self, bond):
self.backupMol()
bondtype = bond.GetBondType()
bondtypes = [Chem.rdchem.BondType.TRIPLE,
Chem.rdchem.BondType.SINGLE,
Chem.rdchem.BondType.DOUBLE,
Chem.rdchem.BondType.TRIPLE]
#Find the next type in the list based on current
#If current is not in list? Then it selects the first and add 1 => SINGLE
newidx = np.argmax(np.array(bondtypes) == bondtype)+1
newtype = bondtypes[newidx]
bond.SetBondType(newtype)
self.molChanged.emit()
Similar approaches are used to toggle R/S and E/Z stereochemistry.
Putting it together in a desktop app
The rdEditor.py brings it all together as a QMainWindow widget with a central widget (molEditWidget), and menus and buttons to select the actions and atom and bond-types. The buttons and menu’s reuse QActions, that bind together an icon, a name, a shortcut key, a status tip and what method to bind. As example for the openAction:
self.openAction = QAction( QIcon(self.pixmappath + 'open.png'), 'O&pen',
self, shortcut=QKeySequence.Open,
statusTip="Open an existing file",
triggered=self.openFile)
This action is then reusable as a button in the toolbar and an entry in the drop-down menu.
self.mainToolBar.addAction(self.openAction)
self.fileMenu.addAction(self.openAction)
The InitGUI method sets some definitions of the main window and also sets the central widget. It also calls the SetupComponents method which takes care of creating all actions and reuse them in menu’s and toolbars. All QActions are created in the CreateActions method. Likewise the menus and toolbars are created with the CreateToolbars and CreateMenus method. A lot of the actions are bound to the same method, which passes on the events senders object name to the method on the molEditWidget. As an example, if a button/menuitem called “P” is bound to the .setAtomType method, the method will pass the sender objects name, “P”, to the setAtomType on the editor widget, which will choose phosphorous as the atom type to use. This simplifies things a lot, as example when the whole periodic system needs to be added in the pTable widget. This can then be done iteratively via some loops instead of defining each qAction and button with its own method, as example:
for key in self.ptable.keys():
atomname = self.ptable[key]["Symbol"]
action = QtWidgets.QAction( '%s'%atomname,
self,
statusTip="Set atomtype to %s"%atomname,
triggered=self.atomtypePush, objectName=atomname,
checkable=True)
self.atomActionGroup.addAction(action)
The ptable was derived from the mendeleev python package and stored in a file.
Some of the selections are naturally exclusive (e.g. atom-types, bond-types and actions). In the code this is handled by defining QActionGroups that are exclusive. As in the example above, the atom QActions are added together in an atomActionGroup in the last line.
Final thoughts
When I started out I thought it would be harder to write the editor, but PySide helps a lot with the logics of the interface and handling events with QSignals and RDKit handled the chemistry and drawing code. I hope that this brief walk-through of the structure and overview of the program code can help as inspiration for other GUI projects or using/modifying the code, which is available at GitHub: http://github.com/EBjerrum/rd-editor
UPDATE 2024-april-02:
There is now a preprint on rdEditor. As there has been some recent interest in the software it will be getting some maintenance and upgrades.